Sie sind hier: Startseite > Nieren > ADPKD
Autosomal dominante polyzystische Nierenerkrankung (ADPKD)
Definition der autosomal dominanten polyzystischen Nierenerkrankung
Die autosomal dominante polyzystische Nierenerkrankung (ADPKD) ist eine häufige monogenetische Multisystemerkrankung mit progredienter Ausbildung multipler Nierenzysten, fortschreitender Einschränkung der Nierenfunktion im Erwachsenenalter, Leberzysten und intrakraniellen Aneurysmen.
Epidemiologie
1:2000. 7–15 % der dialysepflichtigen Patienten haben ADPKD.
Ätiologie der autosomal dominanten polyzystischen Nierenerkrankung
Genetik:
Die meisten Patienten haben pathogene Varianten in zwei Genen: in 85–90% ist PKD1 (Chromosom 16, kodiert Polycystin-1) betroffen, in 10–15% PKD2 (Chromosom 4, kodiert Polycystin-2). PKD2-Mutationen verursachen typischerweise einen milderen Krankheitsverlauf (späterer Beginn, langsamere Progression). Selten verursachen andere Genveränderungen die ADPKD oder einen ADPKD-ähnlichem Phänotyp. Autosomal dominante Vererbung mit sehr hoher Penetranz ist typisch, sodass 50% der Kinder von betroffenen Patienten die Erkrankungen vererbt bekommen. Die Erkrankung entsteht gemäß der Knudson-Theorie der zwei Treffer: ein erkranktes Allel wird vererbt, das zweite Allel wird durch eine spätere spontane Mutation verändert und erklärt die lange symptomfreie Latenz bis zum Erkrankungsbeginn.
Pathophysiologie:
Polycystin-1 und Polycystin-2 erfüllen wichtige Aufgaben in der Signaltransduktion und in der Ausbildung des Primärziliums der Tubulusepithelzelle. Eine Störung führt zu einer Störung der Zellpolarität, Proliferation des Tubulusepithels und zur Ausbildung von Zysten; jeder Nephronabschnitt kann betroffen sein. Ähnliche Mechanismen schädigen die Blutgefäße und andere Organsysteme. Die Erkrankung führt zu einer Aktivierung der mTOR Signaltransduktion, Erhöhung der cAMP-Produktion und Erhöhung der Ionen- und Flüssigkeitssekretion in das Nierenzystenlumen. Der Vasopressin V2-Rezeptor-Blocker Tolvaptan hemmt die ADH-abhängige cAMP-Produktion und verlangsamt die Zunahme des Nierenvolumens und verzögert die Entwicklung einer Niereninsuffizienz.
Progressionsfaktoren:
Das normale (beidseitige) Nierenvolumen beträgt in der Regel unter 400 ml. Bei der ADPKD geht eine zunehmende Nierenvergrößerung mit einer Abnahme der Nierenfunktion einher. Das Nierenvolumen kann entweder über MRT-Protokolle gemessen werden, alternativ genügt auch die sonographische Volumenbestimmung. Je größer das Nierenvolumen, desto schlechter die Prognose für die Nierenfunktion. Die Zeit bis zur terminalen Niereninsuffizienz für einen 30jährigen Mann in Abhängigkeit des Nierenvolumens beträgt beispielsweise 10 Jahre (Nierenvolumen 2000 ml), 18 Jahre (1500 ml) oder 21 Jahre (1000 ml) (Kuehn und Walz, 2015).
Pathologie der autosomal dominanten polyzystischen Nierenerkrankung (ADPKD)
Im Krankheitsverlauf entstehen sehr stark vergrößerte Nieren, welche komplett von Zysten unterschiedlicher Größe durchzogen sind [Abb. polyzystische Niere]. Histologisch stammen Zysten aus verschiedenen Nephronsegmenten. Extrarenal finden sich häufig Leberzysten [Abb. Leberzysten], seltener Pankreaszysten und weitere Manifestationen (siehe Klinik).
Klinik der autosomal dominanten polyzystischen Nierenerkrankung (ADPKD)
Erkrankungsbeginn:
30. bis 50. Lebensjahr. Vermehrtes Screening durch Sonographie und genetische Diagnostik senkt das Alter für die Erstdiagnose.
Beschwerden:
Typische Symptome und Komplikationen sind arterieller Hypertonus, Hämaturie, Proteinurie, Flanken- oder Bauchschmerzen, rezidivierende Zysteninfektionen und Nephrolithiasis.
Assoziierte Fehlbildungen:
Zysten in Leber, Pankreas und anderen Organen. Aneurysmen der Hirnarterien mit Gefahr der Subarachnoidalblutung. Erhöhtes Risiko für Kolondivertikel, Herzklappenvitien oder thorakale Aortenektasie.
Diagnose der autosomal dominanten polyzystischen Nierenerkrankung (ADPKD)
Familienanamnese:
Familienanamnese über drei Generationen mit Erfassung von Nierenversagen, Zystenerkrankungen oder Subarachnoidalblutung.
Labor:
Urinuntersuchung (Proteinurie? Hämaturie?), Kreatinin, bei Beschwerden mit CRP.
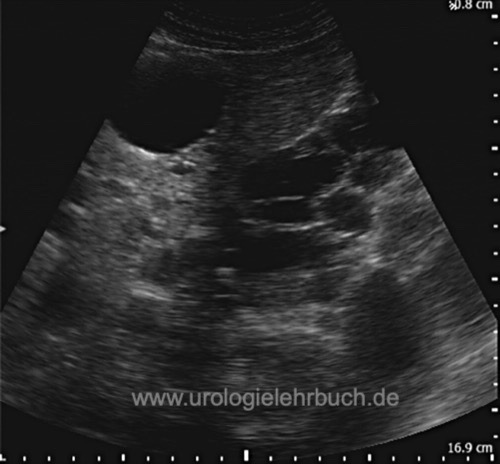
Sonographie:
Bei Erwachsenen mit positiver Familienanamnese ist die Abdomensonographie die empfohlene Erstuntersuchung. Es werden die Nieren, Leber, Pankreas und Milz auf Organzysten untersucht. Die Bestimmung des beidseitigen Nierenvolumens ist wichtig für die Einschätzung der Prognose hinsichtlich einer späteren Niereninsuffizienz bei Manifestation der ADPKD. Altersabhängige Kriterien verbessern die diagnostische Sicherheit: bei 15–39 Jahren sprechen mindestens 3 Nierenzysten insgesamt für ADPKD; bei 40–59 Jahren mindestens 2 Zysten pro Niere; ab 60 Jahren mindestens 4 Zysten pro Niere. Zum Ausschluss einer ADPKD bei positiver Familienanamnese gilt: im Alter von 15–39 Jahren schließt das Fehlen oder Vorliegen von höchstens 1 Zyste die Erkrankung weitgehend aus, im Alter von 40–59 Jahren das Vorliegen von höchstens 2 Zysten insgesamt.
 |
Genetische Diagnostik:
Die genetische Diagnostik ist indiziert zur Differentialdiagnose bei unklaren zystischen Nierenerkrankungen und als prädiktive Diagnostik bei positiver Familienanamnese und unklarer Bildgebung.
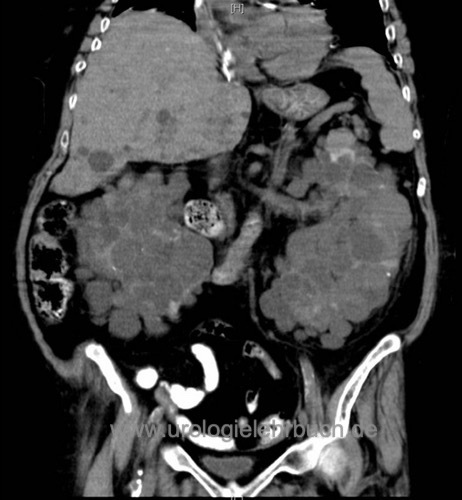
Computertomographie oder MRT:
Die abdominelle Schnittbilddiagnostik ist indiziert bei unklarer Sonographie, zur exakten Volumenbestimmung der Nieren und bei Komplikationen (Blutung, Infektion, Nephrolithiasis). Eine kranielle Bildgebung ist zur Risikoabschätzung bezüglich von Hirnaneurysmen möglich.
 |
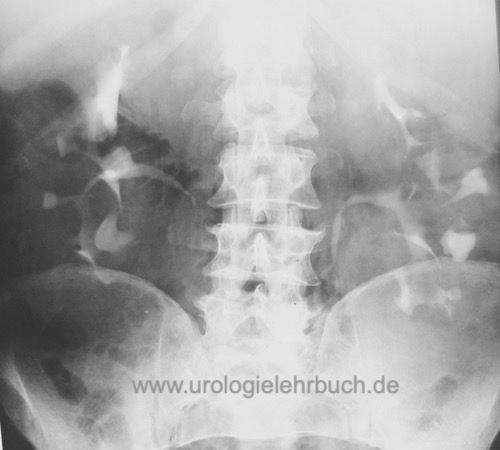
Urogramm:
Ein Urogramm hat heute praktisch keine Bedeutung mehr in der Routinediagnostik der ADPKD. Im Nephrogramm zeigt sich ein "Schweizer Käse"-Aspekt und Kelchverdrängung durch Zysten [Abb. Urogramm bei ADPKD].
 |
Therapie der autosomal dominanten polyzystischen Nierenerkrankung (ADPKD)
Konservative Therapie:
Frühzeitige Therapie des art. Hypertonus mit Medikamenten aus der Klasse der ACE-Hemmer oder der Angiotensin II-Rezeptorantagonisten. Kochsalzarme Diät. Eine Proteinreduktion verbessert nicht die Prognose. Der Vasopressin(V2)-Rezeptorantagonist Tolvaptan ist bei Patienten mit hohem Progressionsrisiko zugelassen, um das Voranschreiten der Niereninsuffizienz zu verlangsamen (Torres u.a., 2012). Tolvaptan verzögert den Beginn der Dialyse um ca. ein Jahr pro fünf Jahre Therapie. Problematisch ist die Polyurie (teilweise über 7 l/d) und die idiosynkratische Hepatotoxizität, dies macht eine intensive Patientenüberwachung notwendig.
Flankenschmerzen:
Nephrolithiasis, Blutung oder Infektion von Zysten können für Flankenschmerzen verantwortlich sein. Bei Zysteninfektion ist eine mehrwöchige Therapie mit lipophilen Antibiotika notwendig. Wenn die Bildgebung veränderte Zysten erkennen lässt, kann eine perkutane Behandlung (Pigtail Kathetereinlage und Sklerotherapie) oder eine laparoskopische Entfernung der Zysten hilfreich sein.
Terminale Niereninsuffizienz:
Hämodialyse, Peritonealdialyse und Nierentransplantation sind die Behandlungsalternativen. Die Nephrektomie der Zystennieren vor Transplantation ist nicht regelhaft erforderlich, sondern nur bei spezifischen Indikationen wie rezidivierender schwerer Infektion, symptomatischer Nephrolithiasis, rezidivierender oder schwerer Zystenblutung, therapierefraktären Schmerzen, Tumorverdacht oder Platzmangel für das Transplantat.
Experimentelle Therapieansätze:
Die medikamentöse Blockierung der Tubulusepithelproliferation durch Inhibitoren der Signaltransduktion wird untersucht. Bisherige Studien mit mTOR Inhibitoren wie Everolimus oder Tyrosinkinasehemmer konnten keinen protektiven Einfluss auf die Nierenfunktion nachweisen.
Prognose der autosomal dominanten polyzystischen Nierenerkrankung (ADPKD)
Die Prognose der ADPKD hat sich in den letzten Jahrzehnten aufgrund besserer Therapieoptionen verbessert.
Niereninsuffizienz:
Das mediane Alter für die terminale Niereninsuffizienz ist abhängig von der ursächlichen Mutation und liegt bei 54–58 Jahren (PDK1 Mutation) und 74–78 Jahren (PDK2 Mutation).
Hirnblutung:
9 % sterben an einer Subarachnoidalblutung (Aneurysmablutung). Zusätzlich sind Hirnblutungen aufgrund einer malignen Hypertonie möglich.
| ARPKD | suchen | Nephronophthisis |
Sachregistersuche: A B C D E F G H I J K L M N O P Q R S T U V W X Y Z
Literatur autosomal dominante polyzystische Nierenerkrankung (ADPKD)
Devuyst O, Torres VE, u. a. KDIGO 2025 Clinical Practice Guideline for the Evaluation, Management, and Treatment of Autosomal Dominant Polycystic Kidney Disease (ADPKD). Kidney International. 2025.
 English Version: autosomal dominant polycystic kidney disease
English Version: autosomal dominant polycystic kidney disease
Urologielehrbuch.de ohne Werbung
Diese Internetseite ermöglicht mit Hilfe von Werbung den Volltext-Zugriff auf das aktuelle Urologielehrbuch.de. Viele Bilder sind zum Schutz von Laien verpixelt oder ausgeblendet. Regelmäßig wiederkehrende (fachkundige) Leser können die Werbebanner abschalten und Zugriff auf alle Abbildungen erhalten: Werden Sie Mitglied über die Crowdfunding-Plattform Steady und unterstützen Sie damit Urologielehrbuch.de.
Urologielehrbuch.de als Hardcover-Buch
 Aktuell, detailliert und übersichtlich: Urologielehrbuch.de wird auch als hochwertiges Hardcover-Buch veröffentlicht. Die 17. Auflage (Ausgabe 2024) ist seit Oktober 2024 verfügbar, siehe Abschnitt Neuigkeiten für die Aktualisierungen und Links für den Buchkauf.
Aktuell, detailliert und übersichtlich: Urologielehrbuch.de wird auch als hochwertiges Hardcover-Buch veröffentlicht. Die 17. Auflage (Ausgabe 2024) ist seit Oktober 2024 verfügbar, siehe Abschnitt Neuigkeiten für die Aktualisierungen und Links für den Buchkauf.