Sie sind hier:Startseite >Nieren >chronische Niereninsuffizienz > Ursachen
Definition und Ursachen der chronischen Niereninsuffizienz
- Chronische Niereninsuffizienz: Ursachen und Folgen
- Chronische Niereninsuffizienz: Klinik, Diagnose und Therapie
Definition der chronischen Niereninsuffizienz
Unter einer chronischen Niereninsuffizienz wird eine dauerhafte Reduktion der glomerulären Filtrationsrate oder das Vorhandensein einer dauerhaften Nierenschädigung verstanden.Abkürzungen und Synonyme: CNI (chronische Niereninsuffizienz), CNV (chronisches Nierenversagen),CKD (chronic kidney disease). In der Definition der KDIGO-Leitlinien werden zwei Bedingungengenannt, eine Bedingung reicht für die Diagnose aus:
- GFR <60 ml/min (normiert auf eine Körperoberfläche von 1,73m2) über 3 Monate, mit oder ohne Nierenschädigung
- Wenn Abweichungen von der normalen Struktur oder Funktion der Nieren längerals drei Monate bestehen und von gesundheitlicher Relevanz sind. Beispiele: Albuminurie,Veränderungen des Urinsediments, Elektrolyt- und andere Veränderungen aufgrund tubulärerStörungen, histologisch nachgewiesene Veränderungen, durch bildgebende Verfahren nachgewiesenestrukturelle Veränderungen oder nach Nierentransplantation.
Stadien der chronischen Niereninsuffizienz:
Die Minderung der glomerulären Filtrationsrate im Rahmen der Niereninsuffizienzwird gemäß der KDIGO-Leitlinien in sechs Stadien eingeteilt [Tab.Stadien der chronischen Niereninsuffizienz].
Stadien der Albuminausscheidung
Die Albuminausscheidung wird in drei Stadien A1–A3 eingeteilt und dient zur weiteren Risikoeinschätzung der Nierenerkrankung, siehe Tab. Stadien der Albuminausscheidung.
Prognose- und Risikoeinschätzung
Die GFR und die Albuminausscheidung hilft bei der Einschätzung der klinischen Bedeutung der Niereninsuffizienz, siehe Tab. Prognose- und Risikoeinschätzung bei chronischer Niereninsuffizienz.
| Stadium | A1 | A2 | A3 | Prävalenz |
| G1 | 55.6% | 1.9% | 0.4% | 58% |
| G2 | 33% | 2.2% | 0.3% | 35% |
| G3a | 3.6% | 0.8% | 0.2% | 4.6% |
| G3a | 1% | 0.4% | 0.2% | 1.6% |
| G4 | 0.2% | 0.1% | 0.1% | 0.4% |
| G5 | 0% | 0% | 0.1% | 0.1% |
| Prävalenz | 93.2% | 5.4% | 1.3% |
Epidemiologie der chronischen Niereninsuffizienz
Die Prävalenz der terminalenNiereninsuffizienz in Deutschland beträgt absolut 60 000, dies entspricht ungefähr 75 pro 100 000. Die Inzidenz der terminalenNiereninsuffizienz beträgt etwa 40 pro 100 000.
Ursachen der chronischen Niereninsuffizienz
Die häufigsten Ursachen für die terminale Niereninsuffizienz in den USA (2008) warendie diabetische Nephropathie (44%), vaskulär-bedingte terminale Niereninsuffizienz(AVK, arterieller Hypertonus) (27%), Glomerulopathien (7%), polyzystische Nierenerkrankungen (3%), tubulo-interstitielle Erkrankungen und andere Ursachen (19%).
Prärenale Ursachen:
Hypovolämie, Sepsis, Schock, Nierenarterienstenose, hepatorenales Syndrom, Herzinsuffizienz (kardiorenales Syndrom).
Glomerulonephritiden:
Primäre Formen der GN, IgA-Nephropathie, membranöse Nephropathie, FSGS, Lupusnephritis, ANCA-assoziierte Vaskulitiden und andere immunologische Erkrankungen.
Interstitielle Nephritis:
Nephrolithiasis, Harnstau, vesikoureteralerReflux, Gicht, Nephrokalzinose, medikamenteninduzierte (allergische) Nephritis, Analgetikanephropathie, Balkannephropathie ausgelöst durch Aristolochiasäure.
Zystische Nierenerkrankungen:
ADPKD, ARPKD, tuberöse Sklerose, Markschwammniere, juvenile Nephronophthise, medulläre zystische Erkrankung, kongenitales Nephrose-Syndrom.
Systemerkrankungen:
Diabetes mellitus, arterielle Hypertonie, Autoimmunerkrankungen und Vaskulitis(Goodpasture-Syndrom, Wegener-Granulomatose, Polyarteriitis nodosa, SLE,Purpura Schönlein-Henoch, thrombotisch thrombozytopenische Purpura), Amyloidose, ...
Mikroangiopathien:
Hämolytisch-urämisches Syndrom, DIC, maligne Hypertonie, Antiphospholipid-Antikörper.
Postrenale Ursachen:
Subvesikale Obstruktion (Harnröhrenklappen, BPH), vesikoureteralerReflux, Prune-belly-Syndrom, Harnleitersteine.
Pathophysiologie der chronischen Niereninsuffizienz
Glomeruläre Adaptation bei Nephronverlust:
Die verbliebenen Nephrone hypertrophieren und steigern die Einzelnephron-GFR. Nach einer Nephrektomie steigt die initiale postoperative GFR innerhalb von wenigen Monaten von 50 % auf 80 % des Ausgangswerts an.
Mechanismen der GFR-Erhöhung: erhöhte glomeruläre Durchblutung mit Erhöhung desglomerulären Plasmaflusses, erhöhter glomerulärer hydrostatischer Druck und Erhöhungder Filtrationsfläche durch Hypertrophie des Glomerulums.
Glomerulotubuläre Balance:
Der Funktionszustand des Tubulus ist einSpiegel der glomerulären Funktion. Zu einem hypertrophierten Glomerulum gehört einhypertrophierter Tubulus, während ein atrophes Glomerulum einen atrophen und obliteriertenTubulus induziert.
Intakte Nephron-Theorie:
Eine fortschreitende chronische Niereninsuffizienz ist meist der Ausdruck einesprogressiven Nephronenverlustes, die restlichen Nephrone sind hypertrophiert und leistenmeist mehr als ein einzelnes Nephron in einer gesunden Niere.
Ab einer gewissen Hyperfunktion entsteht ein chronischer Schaden an diesen verbleibendenNephronen, da die Regenerationskapazitäten überschritten werden. Dies führt zum Fortschreitender Niereninsuffizienz bis zur terminalen Niereninsuffizienz. Histologisches Zeichendes chronischen Nephronschadens ist eine Glomerulosklerose, klinisch entsteht eine Proteinurie.
Ausfall der Ausscheidungsfunktion bei der Niereninsuffizienz:
Die verminderte Urinausscheidung führt zur Akkumulation von körpereigenen Stoffwechselproduktensowie von körperfremden Stoffen (Medikamente). Die höhere Konzentration der Stoffeim Primärharn kompensiert die geringere Nierenleistung und sorgt zunächst für einevergleichbare Ausscheidung (Stadium der kompensierten Retention).
Stoffe mit ausschließlich glomerulärer Filtration (ohne tubuläre Sekretion oderResorption) wie Kreatinin haben Plasmakonzentrationenin direkter Abhängigkeit der GFR. Eine Halbierung der GFR führt zu einer Verdoppelungder Serumkonzentration. Dies bedeutet weiterhin, dass erst ab einer Reduktion derGFR um 75 % eine bedeutsame Konzentrationsänderung im Plasma (vierfach) zu erwartenist. Weitere (geringgradige) Verminderungen der GFR führen aber nun zu dramatischen Änderungen der Kreatininkonzentration [Abb.GFR und Kreatinin].
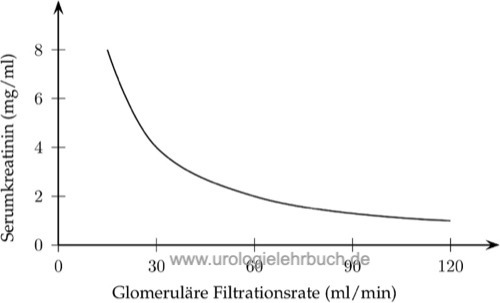
|
Stoffe mit zusätzlicher tubulärer Resorption oder Sekretion können auch bei sehrniedriger GFR normale Konzentrationen im Plasma aufweisen, da über tubuläre Mechanismendie verminderte GFR kompensiert wird (verminderte Rückresorption oder verstärkte Sekretion).Beispiele für diese Elimination sind Phosphat, Urat und Kalium.
Manche Stoffe behalten auch im Endstadium der chronischen Niereninsuffizienz normaleSerumkonzentrationen aufgrund ausgeprägter Kompensationsmechanismen. Beispiele: Natrium und Wasser.
Die Harnstoffausscheidung ist das erste klinische Problem der Retention, die Homöostase des Wasserhaushaltesund der Elektrolyte dekompensiert später. Eine vermehrte Eiweißbelastung oder passagere Verschlechterung der Nierenfunktion lässt die Harnstoffwerte weiterhin ansteigen. Der Entzug des Harnstoffesbei der ersten Dialyse bewirkt ein plötzlichesAbfallen der Diurese.
Störungen des Elektrolythaushaltes bei der Niereninsuffizienz:
Eine GFR über 5 ml/min reicht für einen grenzwertig kompensierten Wasser- und Elektrolythaushalt.Die Urinosmolarität bei einer stark geschädigten Niere liegt nahe am Plasmawert (Isosthenurie), sie kann weder konzentrierten noch stark verdünnten Urin ausscheiden.Die osmotische Harnstoffdiurese führt zu einem gesteigerten Natriumverlust, es kannein Natriummangel mit negativer Auswirkung auf die GFR entstehen.
Eine lebensbedrohliche Hyperkaliämie ist bei der terminalen Niereninsuffizienz nichthäufig, da die Kaliumsekretion im distalen Tubulus intakt ist. Risikofaktoren fürdie Entstehung einer Hyperkaliämie sind: übermäßige Einfuhr, Azidose, Oligurie, Natriummangel, kaliumsparende Diuretika, Hypoaldosteronismus.
Die verminderte Säureausscheidung führt zur Ausbildung einer metabolischen Azidose.
Endokrine Funktionsstörungen bei der Niereninsuffizienz:
Renale Anämie:
Die renale Anämie entsteht aufgrund einer verminderten Erythropoetinsekretion.Renale Osteopathie:
Die fehlende renale Hydroxylierung von 25-Hydroxyvitamin D zu 1,25-HydroxyvitaminD verursacht eine Hypokalziämie und eine Mineralisationsstörung an den Knochen. Weiterhin entsteht ein sekundärerHyperparathyreoidismus.
Sekundärer Hyperparathyreoidismus:
Mit abnehmender GFR wird immer weniger Phosphat ausgeschieden, die steigende Phosphatkonzentrationlässt einen Hyperparathyreoidismus entstehen.
Urämische Funktionsstörungen:
Fast alle Organsysteme werden durch die Urämie geschädigt, dies betrifft vor allemdas Herz- und Kreislaufsystem und erklärt die hohe Mortalität unter Dialyse. Die verantwortlichenToxine bei der Urämie entstammen vor allem dem Proteinstoffwechsel, neben Harnstoff (Urea) sind Guanidine, Urate, Kreatinin, Tryptophan, Tyrosin, Endprodukte des Nukleinstoffwechsel, Polyamine und Phenole zu nennen.
Neben der Entgiftungsfunktion hat die Niere auch eine Inaktivierungsfunktion fürviele endogene Botenstoffe. Eine reduzierte Nierenmasse kann diese Aufgabe nur insuffizientübernehmen (Parathormon, Glukagon, Insulin, LH, Prolaktin...).
Molekulare Mechanismen der Urämie:
Die gestörte Funktion der Na-K-ATPase führt zu Änderungen der elektromechanischen Eigenschaften, des Ruhepotentials sowie des Zellvolumens. Weiterhin ist die verminderte Aktivitätder Na-K-ATPase Ursache für eine Hypothermie und einen verminderten Grundumsatz.
Proteinstoffwechsel:
Die Urämie führt zu einem Protein-und Kalorienmangelzustand mit einer katabolen Stoffwechsellage. Der Gewichtsverlustwird durch die Tendenz der Wasser- und Natriumretention kaschiert.
Kohlenhydratstoffwechsel:
Es besteht eine leichte Glukoseintoleranz, welche per se keine Therapie benötigt(azotämischer Pseudodiabetes). Ursachen sind eine periphere Insulinresistenz und Aktivierung von Glukagon und Katecholamine.
Fettstoffwechsel:
Hypertriglyceridämie und vermindertes HDL-Cholesterin führen zur Arteriosklerose.
Nieren:
Ausbildung einer erworbenen zystischen Nierenerkrankung und Nierentumoren (Nierenzellkarzinom) durchunklare zystogene und mutagene Stoffe der Urämie [siehe Kapitel Erworbene zystische Nierenerkrankung].
| ANV | Suchen | Stadien Niereninsuffizienz |
Sachregistersuche:ABCDEFGHIJKLMNOPQRSTUVWXYZ
Literatur chronische Niereninsuffizienz
Meguid El Nahas und Bello 2005 MEGUIDEL NAHAS, A. ; BELLO, A. K.:
Chronic kidney disease: the global challenge.
In: Lancet
365 (2005), Nr. 9456, S. 331–40
Ruggenenti u.a. 2001 RUGGENENTI, P. ; SCHIEPPATI, A. ; REMUZZI, G.:
Progression, remission, regression of chronic renal diseases.
In: Lancet
357 (2001), Nr. 9268, S. 1601–8
 English Version: chronic kidney disease
English Version: chronic kidney disease
Urologielehrbuch.de ohne Werbung
Diese Internetseite ermöglicht mit Hilfe von Werbung den Volltext-Zugriff auf das aktuelle Urologielehrbuch.de. Viele Bilder sind zum Schutz von Laien verpixelt oder ausgeblendet. Regelmäßig wiederkehrende (fachkundige) Leser können die Werbebanner abschalten und Zugriff auf alle Abbildungen erhalten: Werden Sie Mitglied über die Crowdfunding-Plattform Steady und unterstützen Sie damit Urologielehrbuch.de.
Urologielehrbuch.de als Hardcover-Buch
 Aktuell, detailliert und übersichtlich: Urologielehrbuch.de wird auch als hochwertiges Hardcover-Buch veröffentlicht. Die 17. Auflage (Ausgabe 2024) ist seit Oktober 2024 verfügbar, siehe Abschnitt Neuigkeiten für die Aktualisierungen und Links für den Buchkauf.
Aktuell, detailliert und übersichtlich: Urologielehrbuch.de wird auch als hochwertiges Hardcover-Buch veröffentlicht. Die 17. Auflage (Ausgabe 2024) ist seit Oktober 2024 verfügbar, siehe Abschnitt Neuigkeiten für die Aktualisierungen und Links für den Buchkauf.